Keywords: molecular modeling, molecular mechanics, electronic structure, computer graphics, chemical education.
We might pause to reinforce our observation that the work of the apprentice has begun with the structural formula shorthand, a symbolic representation of the fundamental units of the chemists mental model of the molecule, i.e. atoms, bonds, and electron pairs. As many have noted, this medium is ideally suited to emphasize key features of a chemical process while capable of suppressing any distraction. Rearrangements of these symbolic or linguistic elements in a kind of abstract space (through which chemists communicate) are representations of potential chemical transformations in real space. It is the plausibility and significance of proposed transformations that are at issue here, and these qualities of the representation are the basis for judgment of the competence of the apprentice. However, the ultimate test of those qualities, transformation of substances in the laboratory, is not to be conducted. There are excellent reasons: the ultimate test is likely to be expensive, time demanding, and uncertain of success. Success may be the ultimate test of the validity of the novices suggestion, but it is the ability of the apprentice to manipulate the language of chemistry that is to be judged. One may well (and often) consider the novices speculation to be evidence of a subtle and powerful scientific mind, and still be skeptical of the ultimate outcome of the work proposed.
The symbolic representation of the Baylis-Hillman reaction has so far been entirely adequate to the task of description. Literature reports of the slowness of the reaction, and the difficulty in generating products of specifically desired chirality constitute a problem worthy of consideration: how can the reaction be accelerated, and how can the product chirality be controlled? These aspects of the molecule one geometrical, one energetic are not naturally incorporated into the molecular sketch.
We celebrate (Rouhi 1999) the insight of vant Hoff (and in rough parallel, Le Bel) who are distinguished in the history of chemistry for taking the leap into the three-dimensional representation of molecules with physical models. The tetrahedra of vant Hoff were so simple and elegant a representation and explanation of the phenomena of optical activity and isomer counts, that though there were scoffers offended by the audacity of the young man, his models were generally accepted immediately. The third dimension (3D) was a key, since mirror images of planar objects must be equivalent. The third dimension is of course not absent from the usual first discussions of chirality and chiral recognition the hand in glove and the foot in shoe are analogies depending for their power on tactile experience in the 3D macroscopic world. The sketch however, confined to two dimensions and borrowing only crudely from the code of perspective drawings, is not so effective in conveying the sense and nature of chirality (Hoffmann 1995, sect. 9). It can incorporate reminders of the 3D character of the tetrahedron depth clues but the success of such reminders must depend on the experience in 3D of the user of the sketch.
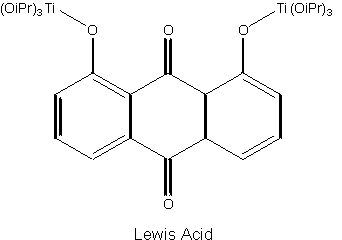
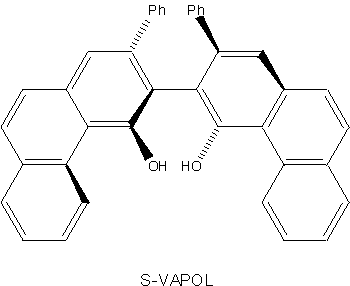
The Lewis acid is to be formed from an achiral titanium species and a chiral vaulted 3,3´-biphenanthrol, which has a two-fold axis of symmetry (not a mirror plane). Here the two-dimensional representation becomes so unwieldy that it begins to lose its utility as a model.
Departures from coplanarity of the two phenanthrene rings are suggested by the bold and broken lines in the diagram of S-VAPOL; atoms at the thick end of the bold wedge are close to the viewer (in front of the plane of representation). Atoms at the end of a broken line are remote from the viewer (behind that plane).
It is left as an impossible exercise, to construct a sketch of the molecule resulting from the attachment of the S-VAPOL to a Titanium atom in the Lewis acid through two oxygen atoms, after splitting out two water molecules.
When a model fails, the next step is often to elaborate the model. Rather than a typical ball-and-stick physical model, one could have constructed a space-filling model. While more realistic in some ways, these objects are generally not as informative as ball-and-stick models, are more difficult to manipulate, and rule out only the conformations in which the spheres assigned to atoms collide. It was at this stage that computer graphics and computer-based (enabled) energy modeling presented itself as an attractive technique.
The appealing feature of computer graphics and associated energy-estimating systems (molecular mechanics, to begin with) is that they promise exactly the information lacking in the abstract [1] model (the sketch) and the iconic model (the physical object). That is an estimate of the relative energy of atomic arrangements, built into an abstract mathematical representation which is expressed on a computer display that has some of the qualities and appeal of the iconic model.[2] But are we to believe such promises? On what foundation do they rest? Moreover, can the computer-contained analogic system be considered a desirable model? We will return to these questions, as our story matures.
This surely seems innocuous, a mere convenience which is more or less equivalent to what the reader of a molecular sketch does. However, it is the first of several significant steps in which a task of interpretation is given over to an inhuman agent that does not carry the wishes and insights of the chemist. Often, early in practice with the drawing system, the intended arrangement of atoms defined in the sketch and its computer-equivalent display, is transformed out of recognition by criteria of the program of what constitutes a stable molecular structure. Giving over the task of interpretation is a surrender of control. The question is of course: what compensation is given?
The result was pleasing in appearance. However, the question hung in the air: if we can impose our own requirements on the model, can we rely on the reports of the computer system? Has it not lost whatever virtue its objectivity, its enforced separation from our hopes and dreams, can bring?
Of course, if the modeling system (however bullied into submission) would still not illustrate the proposal, this question would not need serious response. We can set aside the question for the time.
Molecular mechanics, an analogic model for all its mathematical abstraction, requires a considerable dosage of experimental fact. The quantities (parameters of the model) by which the analogy is realized are evaluated as to match structures and sometimes energies for well-understood systems. The modeling system is calibrated in this way, and one has confidence precisely to the extent that the system under investigation resembles the calibration set. In our case, the analogy is not well tested, the system under investigation is so different from the members of the calibration set, that one should have no exaggerated expectation of its success (or if successful, the significance of its good fortune.) We are in unfamiliar territory, with a guide of little plausibility. But like explorers before us, we press on.
Our hope was realized. That is to say, the S-VAPOL binds to a single titanium, at equatorial and axial positions (a-e binding). With the help of the computer display, we were able to distinguish two kinds of a-e binding, and to believe the systems report that one was much more stable. The bridging alternative is much less favored by our modeling system, and a second S-VAPOL is hard to introduce into the Lewis acid.
With a plausible structure for a chiral catalyst, the next step is to characterize the binding of the substrate aldehyde. This is the species to which a new carbon center is to be defined, with, so one is hoping, controlled chirality. The only chirality in the modeled system thus far is in the Lewis acid catalyst, but its influence on the potentially chiral (prochiral) substrate is critical to the success of the proposal.
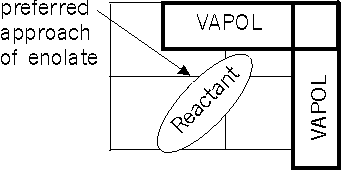
A great simplification of the catalyst topography can be helpful here. The rectangular figure shows a caricature [3] which takes us back from the complexity of the analogic model to a more immediately visualized representation. The rectangle that encloses our entire molecule is bisected twice, by a horizontal line and a vertical line. Our line of sight toward the Lewis acid passes through their intersection. The titanium atoms lie to the left and right of the intersection along the horizontal line, which defines the plane of the anthroquinone. The line of sight is the way the C=O bond of a reactant aldehyde might coordinate with the vacant Titanium coordination sites of the Lewis acid.
Once again we intervened in the process of modeling, since the system
itself refused to coordinate the C=O oxygen along the axis so simple to
define in a sketch. We aligned it by imposing constraints, i.e.
our will. The question already posed (if we can impose our own requirements
on the model, can we rely on the reports of the computer system?) troubled
us somewhat less than it had, for reasons easy to understand (we were hot
on the chase) but not so easy to defend.
We needed to judge the hospitality of the aldehyde (as bound to the chiral catalyst) to an approaching enolate. To do this we tethered an enolate 5 Å away from the carbon center to which it would be eventually attached. Depending on our starting point, we found that under this constraint the enolate migrated into the upper left quadrant. The separation of 5 Å was sufficient that no bond would be formed even in a realistic model capable of description of such an event. The energy field, defined by the van der Waals and electrostatic charges well incorporated into the molecular mechanics description would act as a long-range guide to the enolates approach. We argued that this would determine the course of the reaction long before arrival at the critical site of the transition state.
In this way, we established to our satisfaction that S-VAPOL ligand blocks approach from the upper right quadrant of the figure. It seemed that the ligands bulk also forces the bulky substituent of the attached aldehyde away from that quadrant. This exposes one face of the aldehyde to the final reagent, a phosphine-activated enolate (methacrylate in the model).
Here is the way the results of this shady practice appeared in the research proposal.
There is evidence for reversal of the favored path, from backside attack in polar medium, to topside attack in nonpolar medium. In the terse expression of the technical report:
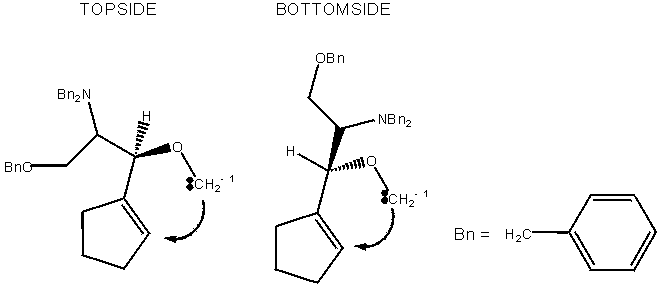
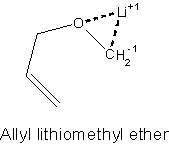
We had available results of a prior electronic structure modeling of the Still-Wittig transition state for a much smaller system, allyl lithomethyl ether (Wu & Houk 1990). We were easily able to reproduce the published structure of this small system, a tribute to advances in computer capability and of course to those who showed the way. We quickly came to agreement with that report that the Lithium counter-ion (though not shown in the first sketch above) was intimately involved in the Still-Wittig transition state.
That is, try as we might to construct a transition state of the type the investigators had drawn, we were unable to enforce our wishes. We could constrain the anion to a form where the new bond from the carbanion site (CH2-1) to the unsaturated C=C terminus was not completely established but was beginning to close. However, once we relaxed constraints, the system collapsed to complete the bond. With lithium present, we could locate transition states with ease. This behavior strengthened our confidence in the model, simply because it was not infinitely malleable; it had principles it would not abandon. Even better, the computational system, left to its own devices, did report that the transition states in which the Li cation is bound to N and O favored a topside arrangements while in the transition states where the Li cation is coordinated to the ether oxygen and a solvent oxygen, the bottomside approach is favored. Simply because we took no measures to impose our wishes on the system, apart from offering for judgment some structures we had drawn, the system fulfilled its role as impartial evaluator.
Unfortunately for the repute of the molecular modeling related in our story, one can hardly say that the system is memorable, i.e., easy to hold in mind. In fact, to the extent that the manipulations in the computer code are withheld from the user, the system declines to be held in mind. (Perhaps the results especially the graphic presentations as shown are striking, but that is another story, about a version of the iconic modeling borrowed for the purpose of display.) The elaborate calibration and parametrization (the preoccupation only of specialists), inescapable parts of modeling systems, must mean that the system is not simple in any fundamental way. Perhaps the user interface makes the choices seem simple, but that too is another story. Whether the model is self-consistent is hard for the user to appreciate. That property can be revealed by paradoxes in reports, but only in the same unfortunate way that the collapse of a bridge reveals error in design.
The user can come to a more reliable judgment on how powerful the modeling system might be, but even here pitfalls abound. One may require that the modeling system be faithful to known results (even those which were counter-intuitive and for which other representations failed), and report aspects of chemical systems which are independently verifiable. This is of course scientific investigation compactly defined, and may be the justification for the modeling in our story.
The design of the software system defines its flexibility; some systems permit the user to augment or alter the parameter set, so to treat unusual atoms, bond types, etc. Others block all modification, for good or ill. Our system permits such modification, and also accommodates atoms by an interesting process of defaults. Every atom is assigned a radius, and the number of linked neighbors will define ideal bond angles for atoms not otherwise defined in local geometry.[4] The system also draws analogies, and estimates a missing parameter defining the stiffness of a valence angle bend X-Y-Z by searching its data base for X-Y-* (* arbitrary). This process is risky, and is properly disclosed in documentation and in a brief message to the user to the effect that a default bending parameter was used. As we saw, defining constraints extends the range of chemical species and behavior treatable in the system. However, the recommended attitude toward these estimates is distrust, and the message should be treated as a warning to be taken seriously.
One might judge the value of a model by its contribution to the everyday thought of a practitioner. Every chemist is fluent in the abstract language of structural formulas; some convey only pedestrian thoughts, while others express their genius. A paper napkin can hold a stroke of insight in a sketch, if not so simple and pregnant with implication as F=ma or E=mc2. But the napkin, and the symbols thereon, are primarily an expression of the chemists judgment and not much of a corrective. While not as common as pencil and paper, computer assisted modeling is accessible to most chemists. As this trend becomes more pronounced, perhaps we will be able to say that the balance will shift, and that a part of the judgment of the expert chemist will come to reside in the machinery rather than the person. With due respect to the knotty issues raised in Searles Chinese-translating system, we might be unsurprised at this continuation of a trend begun with the stereomodels of vant Hoff and le Bel.
Computer models do not seem to fare so well in the criteria for an ideally valuable system, but its persuasive power is considerable. Our apprentice was able to produce a picture a summary of all the labor of the exercise and present it as (first) trustworthy and (second) almost obvious in its implications. Why did it strengthen her argument, in view of the possibilities that the system was inadequate to the specific task, and that on several occasions we imposed our ideas on the modeling system with its acquiescence but against its built-in principles?
We will certainly not bid farewell to the sketch or the ball-and-stick model, or the structural sketch. They were not equal to the task of helping us visualize the subtleties of the three-dimensional arrangements of atoms in the chiral Lewis acid catalyst and its participation in the Baylis-Hillman reaction or the Still-Wittig systems behavior in various media. However, they were capable of posing a question with enough precision that it could be delegated, to variable degree, to the repository of chemical judgment that is the modeling system. Our apprentices will not hesitate to turn to modeling the next time an argument needs buttressing, a speculation needs test, or a question seems beyond experimental reach.
[2] Clearly the computer modeling software constructs an experience in which the abstract, iconic, and analogic aspects of modeling are all present. However the system succeeds or fails depending on the quality of the analogy it provides.
[3] The sketch discussed above is certainly nothing like a structural formula. It is a more simplified representation of only a few aspects of the chemical system, in the spirit of the block diagrams introduced in studies of self-assembly or the more venerable cartoons representing secondary and tertiary structure of polypeptides etc.
[4] For example the geometry of familiar HOH is explicitly modeled to produce bond angles close to the observed value of 105° , but the angle for O-La-O is defined as 180° , simply by the number of neighbors.
Bode, M.; Kaye, P.: 1991, A Kinetic and Mechanistic Study of the Baylis-Hillman Reaction, Tetrahedron Letters, 32, 5611.Hart, S.; Etzkorn, F.: 1999, Solvent-Mediated Stereoselectivity in the Still-Wittig rearrangement, manuscript in draft, University of Virginia Chemistry department.
Hoffmann, R.: 1995, The Same and Not the Same, Columbia Univ. Pr., New York.
Rouhi, A.M.: 1999, Tetrahedral Carbon Redux, Chemical and Engineering News, Sept. 6, 28-32.
Trindle C.: 1984, The Hierarchy of Models in Chemistry, Croatica Chemica Acta, 57, 1231.
Trindle, C.: 1989, Viewing the Molecular Display: Hints from the Artificial Intelligence of Vision, Computers in Chemistry, 13, 159-164.
van Divender, E.A.: 1999, A Catalytic, Asymmetric Baylis-Hillman Reaction Based on a Chiral Bidentate Bis-Titanium Lewis Acid Catalyst, unpublished paper presented to the faculty of chemistry at the University of Virginia in partial fulfillment of the requirements for the degree doctor of philosophy, February 1999.
Wu, Y.-D.; Houk, K. N.; Marshall, J. A.: 1990, Transition Structure for the [2,3]-Wittig Rearrangement and Analysis of Stereochemistry, Journal of Organic Chemistry, 55, 1421.
Carl Trindle:
Dept. of Chemistry, University of Virginia, Charlottesville, VA 22901, USA; cot@virginia.edu